
(α-Gal A). It is caused by mutations in the GLA gene. This gene is in turn responsible for providing instructions for
the formation of α-Gal A enzyme.
● Anderson-Fabry disease
● Angiokeratoma Corporis Diffusum
● Angiokeratoma Diffuse
● Ceramide Trihexosidase Deficiency
● GLA deficiency
● Hereditary Dystopic Lipidosis
Type 1 classic phenotype
Type 2 later-onset phenotype
prevalence is unknown. Data emerging from the newborn screening studies suggest that the incidence of Fabry
disease varies in different geographic regions. The type 2 later-onset phenotype is more frequent than the type 1
phenotype.
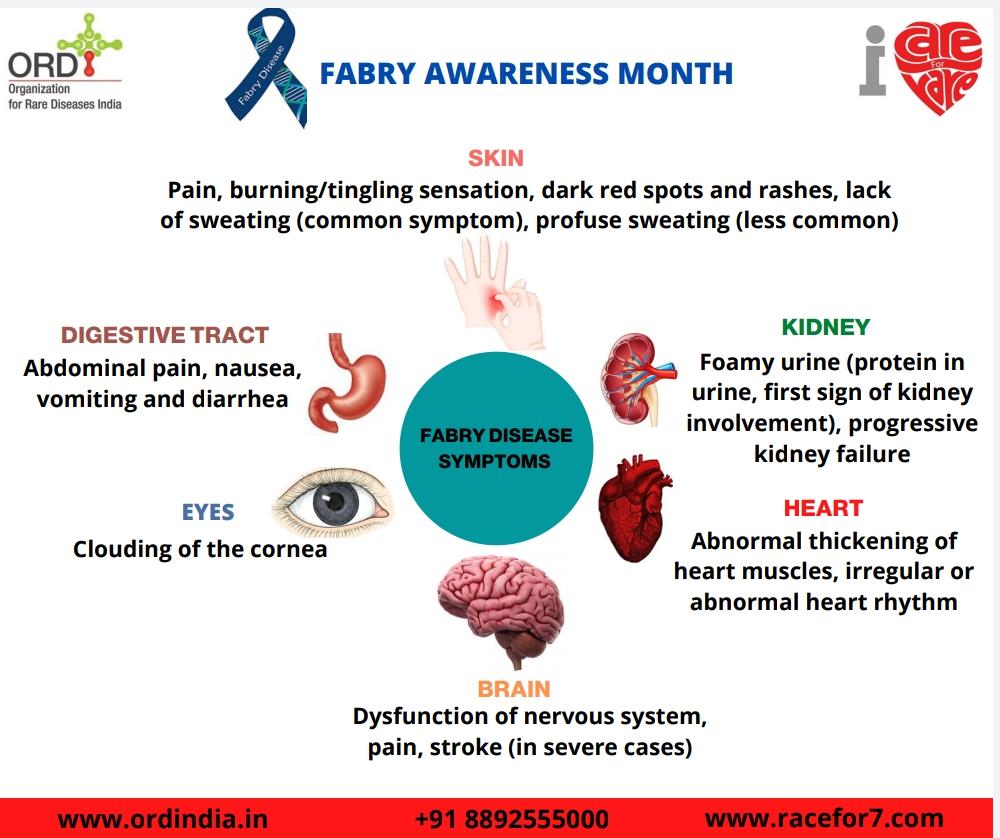
| 1. Pain and Acroparesthesia | Pain is an early symptom and may occur as early as 2-8 years of age. Affected
individuals may experience episodes of severe burning pain in the hands and the feet ( acroparesthesia ). Severe episodes of pain ( Fabry’s crises ) may last for hours to days and are frequently triggered by exercise, fatigue, stress, and/or fever. |
| 2. Anhidrosis or Hypohidrosis | Type 1 males and some type 1 females have decreased or absent sweat
production (hypohidrosis or anhidrosis). |
| 3. Angiokeratomas | Early symptoms include the appearance of a reddish to dark-blue skin rash,
especially in the area between the hips and the knees. |
| 4. Gastrointestinal problems | Abdominal cramping, frequent bowel movements, and diarrhea may also occur,
particularly after a large meal |
| 5. Corneal dystrophy | Cloudy corneas resulting in a characteristic whorl-like opacity. These changes do
not affect vision. |
| 6. Other symptoms | Chronic fatigue, dizziness, headache, generalized weakness, nausea, and/or
vomiting, delayed puberty, lack of or sparse hair growth, and rarely malformation of the joints of the fingers. Ear damage and progressive organ damage (especially kidney and heart). |
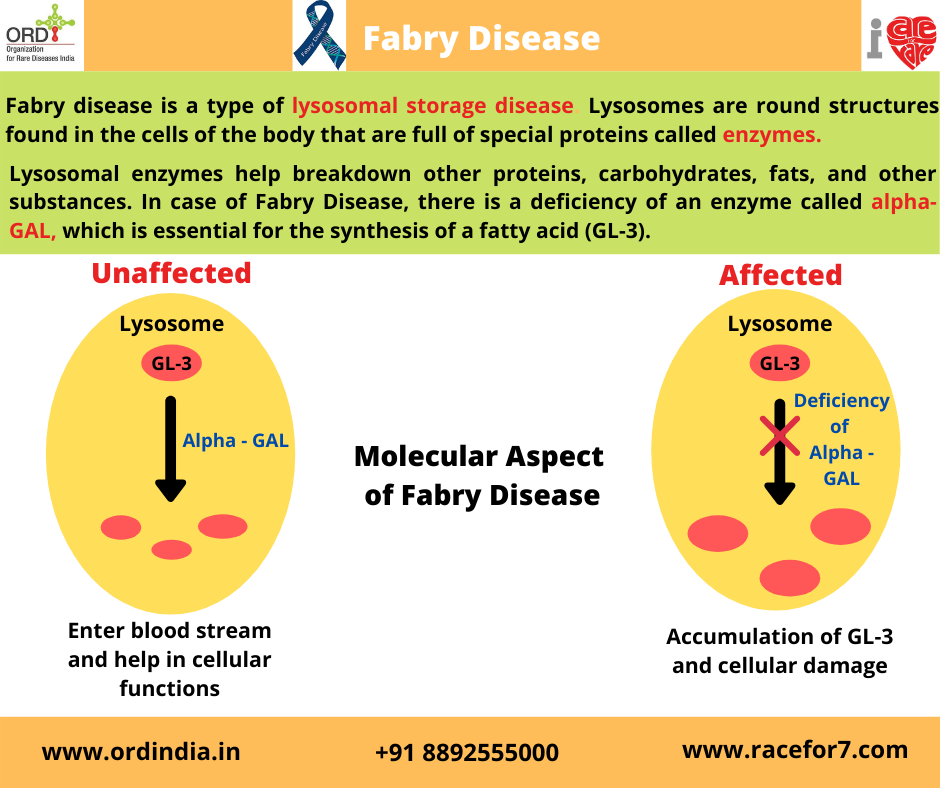
Causes
In Fabry disease, the production of an enzyme called alpha-galactosidase A is affected. This enzyme is active in
lysosomes, which are structures that serve as recycling centers within cells. Alpha-galactosidase A normally breaks
down a fatty substance called globotriaosylceramide. Mutations in the GLA gene alter the structure and function of
the enzyme, preventing it from breaking down this substance effectively. As a result, globotriaosylceramide builds up
in cells throughout the body, particularly cells lining blood vessels in the skin and cells in the kidneys, heart, and
nervous system.
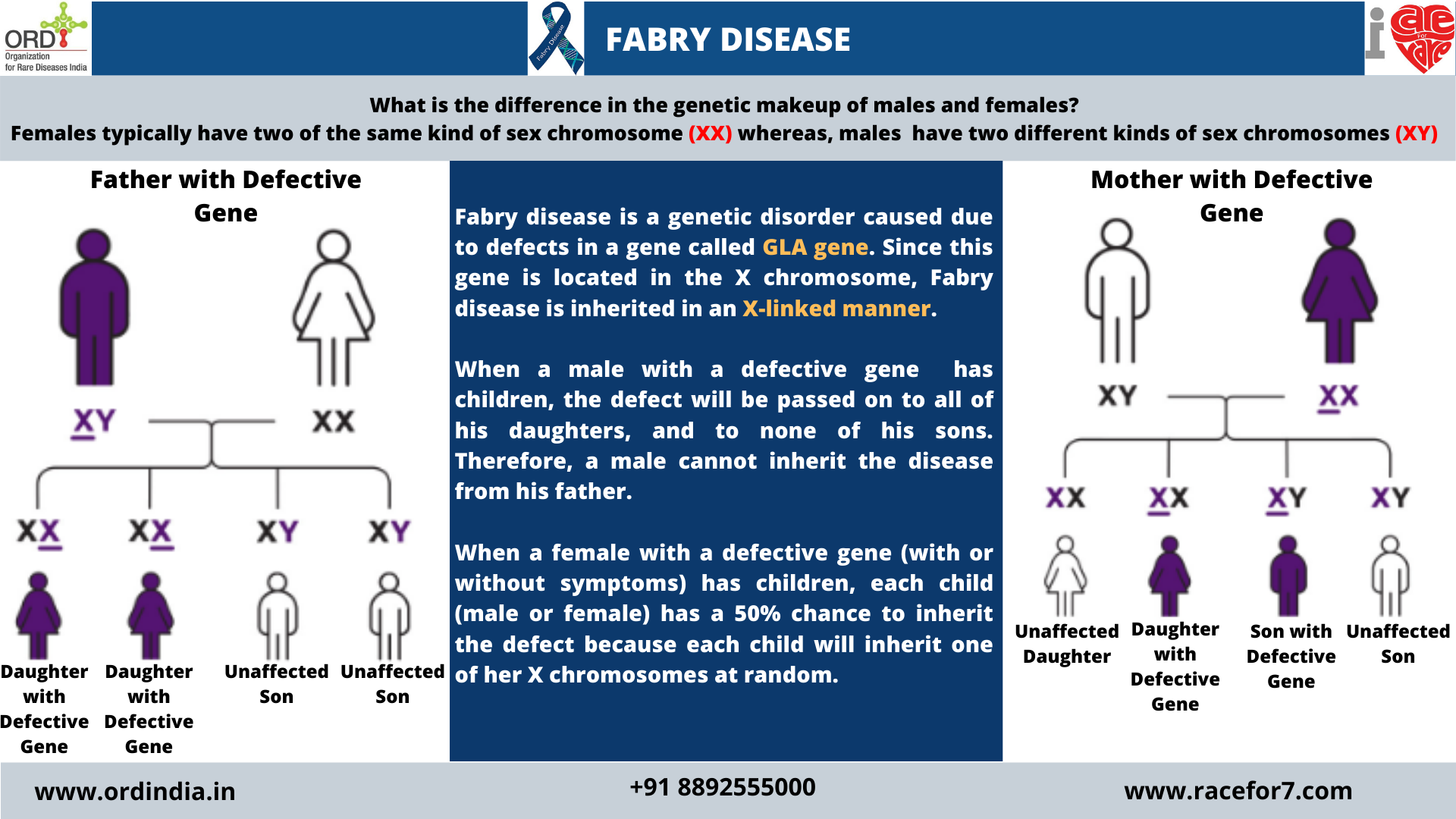
Inheritance Pattern
Since the GLA gene is located in the X chromosome , Fabry disease is inherited in an X-linked manner. When a
male with a defective gene has children, the defect will be passed on to all of his daughters, and to none of his sons.
Therefore, a male cannot inherit the disease from his father. When a female with a defective gene (with or without
symptoms) has children, each child (male or female) has a 50% chance to inherit the defect because each child will
inherit one of her X chromosomes at random.
Diagnosis
● Genetic Testing: Preimplantation (embryo before uterine implantation) genetic diagnosis is available when
the familial mutation in the GLA gene is known.
● Chronic Villus Sampling is a prenatal test that is used to detect birth defects, genetic diseases, and other
problems during pregnancy. During the test, a small sample of cells (called chorionic villi) is taken from the
placenta where it attaches to the wall of the uterus . Early prenatal diagnosis at about 10 weeks of pregnancy
can be made by α-Gal A enzyme and GLA mutation analyses of villi obtained by chronic villus sampling, or
by amniocentesis at about 15 weeks of gestation.
● Clinical Diagnosis of type 1 classic phenotype can be made clinically by physicians who recognize the
characteristic findings of episodic pain in the extremities, absent or decreased sweating (anhidrosis or
hypohidrosis), typical skin lesions (angiokeratoma), gastrointestinal abnormalities, and the corneal dystrophy
in childhood or adolescence.
● Newborn screening studies have identified affected males by demonstrating the reduced α-Gal A activity in
dried blood spots followed by GLA gene sequencing.
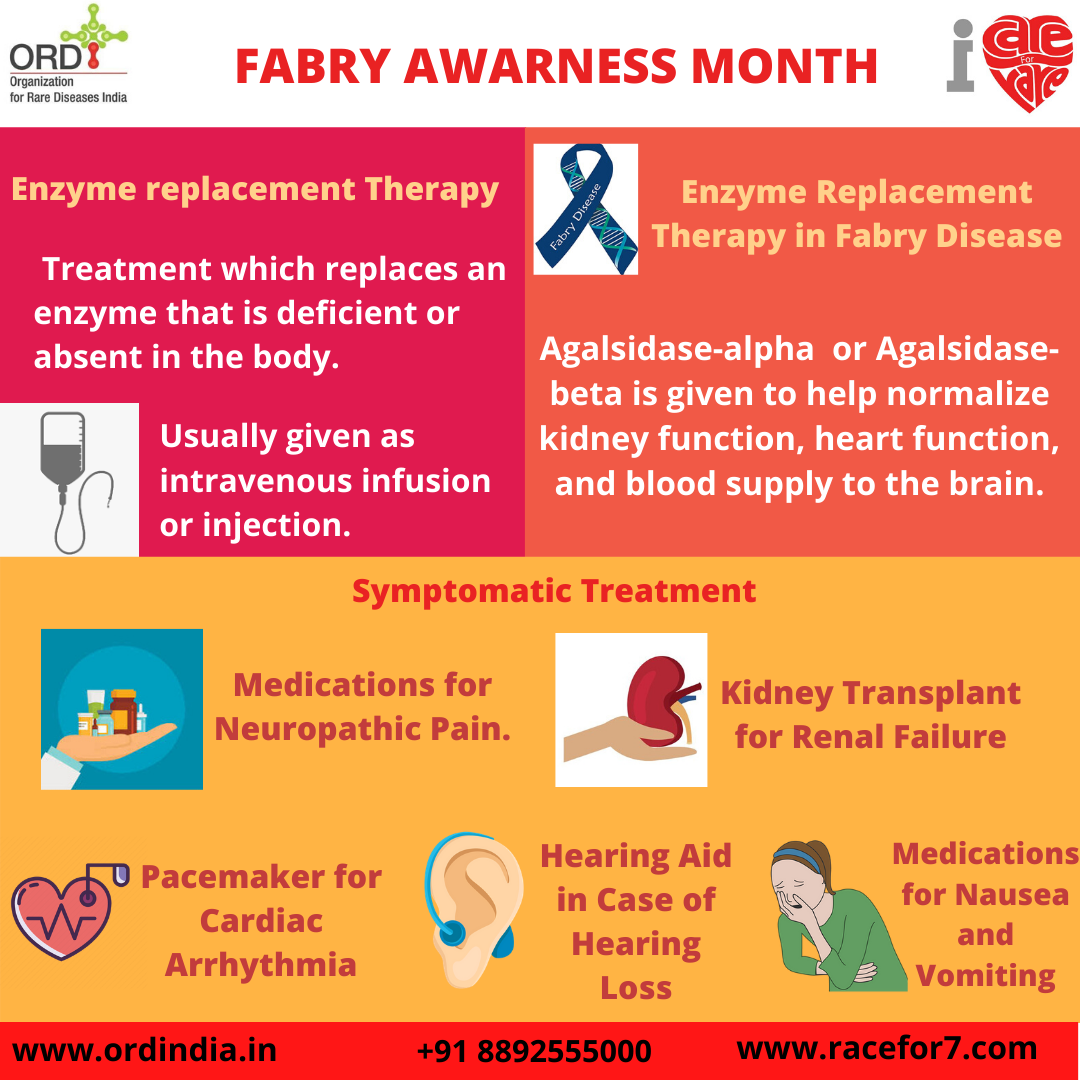
Treatment
Major treatment followed is the Enzyme Replacement Therapy. It is the treatment that replaces an enzyme that is
deficient or absent in the body. Usually given as intravenous infusion or injection. In the case of Fabry disease
Agalsidase-alpha or Agalsidase- beta is given to help normalize kidney function, heart function, and blood supply
to the brain.
Symptomatic treatment includes medications for neuropathic pain, hearing aid in case of hearing loss, medications
for nausea, and vomiting. Other later complications (e.g., kidney failure or heart problems) should be treated
symptomatically after consultation with a physician who is experienced in the care of patients with Fabry disease.
Genetic counseling is recommended for affected individuals and their families.
References
https://rarediseases.org/rare-diseases/fabry-disease /
https://www.medicinenet.com/fabrys_disease/article.htm
Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M; European FOS Investigators. Natural history of
Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006 Apr;43(4):347-52. Epub 2005 Oct 14


